
Un nouveau variant du coronavirus responsable d’un Syndrome Respiratoire Aigu Sévère a provoqué et provoque encore des décès sur tous les continents et dans tous les pays. Ce virus est baptisé Sras-Cov-2 (ou Sars-Cov-2 en anglais) pour évoquer le second variant du premier virus responsable du SRAS en 2003 nommé Sras-Cov. Ces deux espèces virales engendrent un syndrome commun : le SRAS. Mais l’OMS a préféré créer la COVID-19 (COronaVIrus Disease en 2019), soit LA «maladie à Coronavirus 2019 ».
La précision remarquable de cette définition balaye tout risque de confusion, c’est pour cette raison que la gestion de la crise a été exemplaire dans l’ensemble des pays. Sérieusement le Sras-Cov2 provoque la même syndrome que le Sras-Cov, mais de manière plus frustre. Le Sras-Cov-2 est beaucoup plus contagieux, en particulier parce que sa virulence est atténuée. C’est-à-dire que 50 % des personnes contaminantes ne présentent aucun symptôme, donc l’identification et la prise en charge des relais de la propagation du virus sont trop tardives. Je pense que nous pouvons parfaitement mesurer aujourd’hui le poids de ce « trop tard ».
La nature n’a pas attendu l’autorisation de l’OMS pour mettre un nouveau virus en circulation, chez l’Homme. Pour éviter d’intervenir trop tard dans l’avenir essayons de remonter aux origines de l’émergence du « Sras-Cov-2 ». Pour cela nous proposons de tenter de déchiffrer le livre génétique du virus.
Je tiens à te prévenir cher lecteur que si tu attends une réponse définitive, tu seras immanquablement déçu. Le propos de cet article n’est pas nécessairement d’arriver à destination mais de réaliser un voyage dans le monde de la biologie moléculaire où le binaire immuable n’existe pas.
Sommaire
- Notions d’information génétique
- D’où viennent des coronavirus?
- Etude de la protéine spike
- Le Devenir du Sras-Cov-2.
- Conclusions
Notions d’information Génétique
Le génome du Sras-Cov 2 ressemble au génome de la plupart des Coronavirus. Le génome est une séquence (liste ordonnée) de nucléotides (Cf fig1), de la même manière qu’une information numérique est une séquence de bits. Mais lorsqu’un bit peut prendre une valeur de 1 ou 0, un nucléotide peut prendre 4 valeurs : A, G, C, T. Dans le cas d’un coronavirus le génome est constitué par un simple brin d’ARN, donc la Thymine est remplacée par un Uracile soit T=U.
Par ailleurs un octet est une séquence de 8 bits qui représentait l’unité de l’information des premiers microprocesseurs. Aujourd’hui cette unité comporte plutôt 64 bits. Du côté des êtres vivants l’unité d’information est le codon composé de trois nucléotides. Un triplet de nucléotides permet de coder 64 acides aminés différents. Cependant une protéine ne contient que 20 types d’acides aminées conventionnels. Ainsi le code génétique de traduction des bases nucléotidiques en acides aminées est redondant ou dégénéré. Cette propriété :
- permet de déterminer une fréquence relative des acides aminés. Par exemple la leucine est très courante parce qu’elle est traduite à partir de 6 codons différents mais synonymes. A l’inverse le tryptophane et la méthionine ne sont représentés que par un seul codon chacun. La méthionine est particulière car elle constitue le premier acide aminé d’une chaîne protéique, elle correspond au codon d’initiation (Voir Figure 1).
- assure un contexte syntaxique propice aux mutations en permettant des mutations silencieuses et non silencieuses asynchrones sur un même codon, mais aussi en apportant une contrainte sur les mutations non silencieuses qui limite les mutations létales (Voir Figure 2).
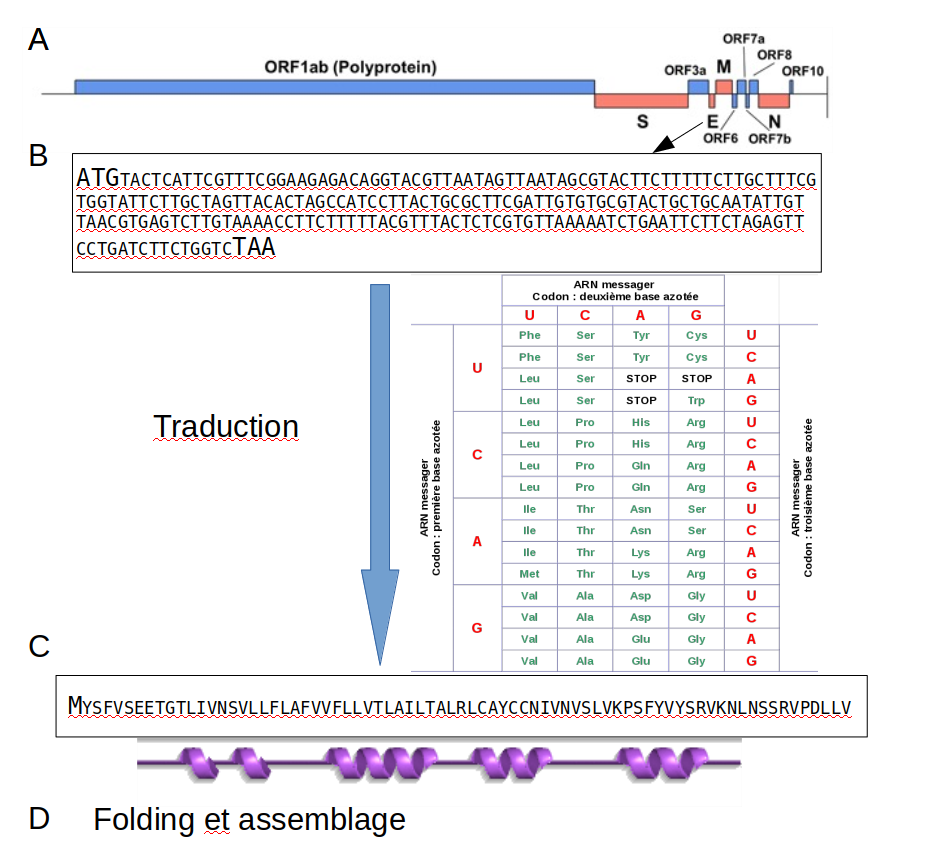
Figure1: Du code génétique à la fonction
(A) Le génome des coronavirus contient environ 30000 nucléotides, c’est le plus long génome parmi les virus à ARN. Il permet de fabriquer des protéines structurales (en rouge) à l’origine de la morphologie de la particule virale (virion). Egalement des protéines non structurales (en bleu) sont produites et sont capables d’interférer avec le métabolisme de l’hôte.
(B) La traduction du code génétique du gène E pris en exemple ici en une protéine dite d’enveloppe nécessite une table de traduction du codon vers l’acide aminé, mais aussi un cadre de lecture (ORF). Ce cadre débute par un codon d’initiation codant pour une méthionine et se termine par un codon STOP. Un décalage du cadre de lecture d’un nucléotide entraîne la production d’une protéine totalement différente. Cette stratégie de compression de l’information est utilisée par les Morbillivirus (ex : rougeole, maladie de Carré) dont la longueur du génome est deux fois moins importante que celle des coronavirus. L’ORF1ab du coronavirus est un peu particulier puisqu’il est traduit en une longue poly-protéine qui est ensuite scindée par des protéases virales en 27 protéines ayant des fonctions différentes mais qui participent toutes à la réplication du virus et donc au contrôle des mutations.
(C) La séquence d’acides aminés constitue la structure primaire de la protéine et elle détermine sa structure tridimensionnelle future. La contiguité de certains acides aminés permet d’établir des liaisons hydrogènes, entre les atomes du squelette de la protéine, à l’origine d’une structure secondaire représentée ici par des hélices alpha.
(D) La structure tertiaire correspond à la conformation spatiale tri-dimentionnelle acquise par la protéine au cours d’un processus de repliement (folding) dirigé par des contraintes énergétiques inter-atomiques et qui aboutit à la conformation la plus stable dans l’environnement du moment. Cette conformation correspond à une situation d’équilibre thermodynamique qui peut changer lors de l’application de contraintes externes. Enfin la protéine monomérique peut encore s’assembler en un polymère plus stable et fonctionnel. Dans le cas présent, le complexe de protéine d’enveloppe du virus est un pentamère organisé en un canal ionique qui s’insère dans la membrane virale.
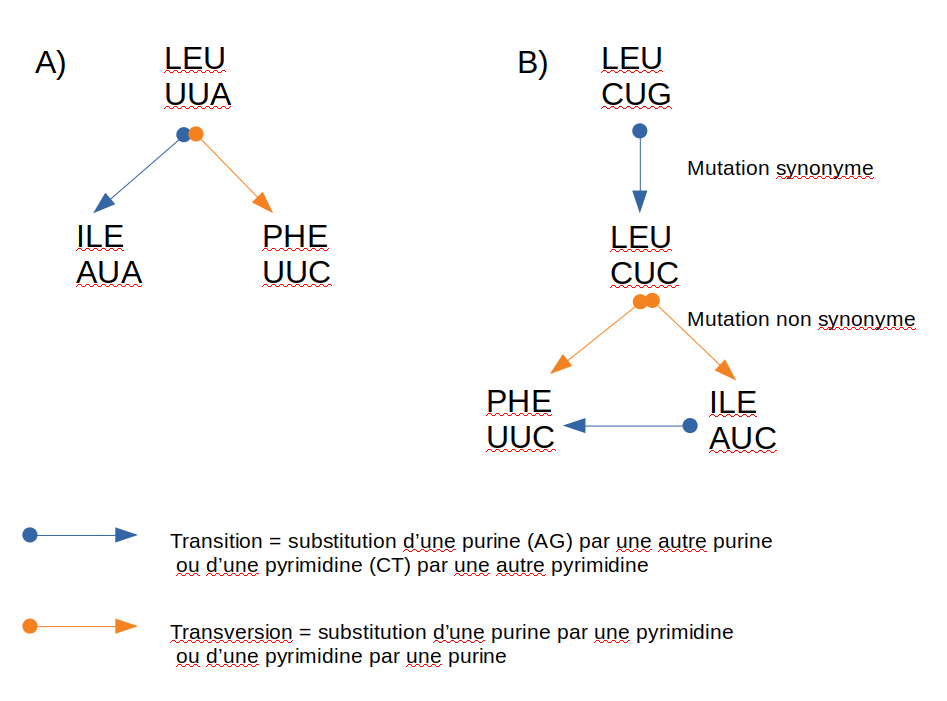
Figure 2: Mutation synonyme vs mutation non synonyme.
La leucine (LEU), l’isoleucine (ILE) et la phénylalanine (PHE) sont des acides aminés hydrophobes (ou non polaires). Ces acides aminés ont tendance à s’enfouir au sein de la protéine et sont rarement exposés à l’extérieur de la protéine au contact d’un milieu aqueu. En revanche ils présentent une forte affinité pour les membranes lipidiques. Ces acides aminés partagent des propriétés physico-chimiques et sont donc relativement substituables dans une protéine.
(A) Pour passer d’une LEU à une ILE, une seule mutation d’un nucléotide Uracile en Adénine est nécessaire. Cette mutation est une transition et elle est peu coûteuse. En revanche pour une transformation de la LEU en PHE une transversion moins probable est requise.
(B) Maintenant si nous considérons un code synonyme de la LEU, le passage vers l’ILE ou la PHE nécessite deux mutations nucléotidiques. La probabilité d’apparition de deux mutations contiguës et contemporaines est hautement improbable. En revanche l’existence des codons synonymes permet une étape intermédiaire du mutation synonyme. Le chemin de la transformation est cependant plus long et plus coûteux que dans le cas précédent (A). Ceci illustre la notion de codons préférentiels qui permettent de poser des contraintes de stabilité plus forte sur certains acides aminés que l’on appelle aussi résidus.
Si vous avez appris quelque chose lors de la lecture de ce premier chapitre, soyez heureux car vous disposez maintenant du bagage nécessaire pour remonter aux sources du virus.
D’où viennent les coronavirus ?
1. Notion de réservoir épidémiologique
Le réservoir d’un virus zoonotique est une « entité assurant la conservation d’un agent pathogène biologique, considéré en tant qu’espèce, et sa fourniture aux sujets réceptifs »[ref]. Les prérequis habituels pour un bon réservoir sont donc :
- La capacité à abriter le virus en autorisant sa multiplication mais en limitant son effet pathogène. Cette cohabitation est le fruit d’une longue co-évolution.
- La capacité à disséminer le virus. L’excrétion du virus dans les fécès et le vol sont très efficaces.
L’histoire de la chauve-souris sur Terre remonte à plus de 50 millions d’années. La paléontologie rapporte qu’à la fin de l’Eocène il existe déjà 26 sous-ordres de chauve-souris. Cette diversité est considérée par les paléontologues comme unique chez les mammifères [ref]. D’ailleurs aujourd’hui les chauves-souris (ordre des Chiroptères) représentent 20 % des 4800 espèces de mammifères.
La chauve-souris est déjà connue comme réservoir naturel de la rage, d’Ebola, de Nipah, de Hendra et du virus de la grippe. Une étude de 2017 menée en Chine [ref] révèle l’existence de 73 espèces de coronavirus à partir d’un échantillon de 1067 chauve-souris appartenant à 21 espèces différentes. En 2009, 103 espèces de coronavirus étaient déjà isolées chez les chauves-souris dans le monde [ref]. Les oiseaux sont considérés comme l’autre réservoir des coronavirus (gamma et delta) , mais aujourd’hui la source incontestée des Bétacoronavirus est la chauve-souris. Leur aptitude au vol (seul mammifère pratiquant le vol propulsé), comme les oiseaux, participe probablement à leur résistance naturelle aux infections à coronavirus. Le métabolisme intense avec forte consommation d’oxygène pendant le vol est à l’origine de la production de dérivés réactifs de l’oxygène qui inhiberaient les réplications virales. Il est à noter que certains coronavirus sont pathogènes chez la poule et la dinde qui ont perdu la capacité de voler. De plus, les chauves-souris produisent de manière constitutive d’importantes quantités d’interféron de type I et III et sont donc insensibles à l’inhibition de la production d’interféron par les coronavirus. Enfin plusieurs voies métaboliques du déclenchement de l’inflammation sont atténuées chez la chauve-souris [ref], ce qui les protège de l’orage cytokinique viro-induit [ref].
A l’inverse l’histoire des hominidés remonte à 7 millions d’années. Une quinzaine d’espèces a été identifiée, mais aujourd’hui une seule espèce persiste et son apparition date de 200 000 ans. Nous pouvons méditer sur ces deux stratégies évolutives opposées : diversification vs exclusivisme.
D’un point de vue pragmatique, les chauve-souris se sont adaptées aux coronavirus au cours de millions d’années, mais pas l’homme moderne, ce qui explique sa vulnérabilité.
2. La classification des coronavirus
La famille des Coronaviridae est extrêmement diversifiée [ref]. La sous-famille des Orthocoronavirinae comprend aujourd’hui 4 genres :
- Les alphacoronavirus se déclinent en 14 sous-genres comprenant, entre autres, les coronavirus félin et canin, le coronavirus de la gastro-entérite transmissible porcine et 2 virus humains qui provoquent le rhume.
- Les bétacoronavirus forment un groupe où se distinguent 5 sous-genres :
- Embecovirus
- Hibecovirus
- Merbecovirus
- Nobecovirus
- Sarbecovirus
- Les gammacoronavirus répandus chez les oiseaux. Pendant très longtemps il comprenait un seul représentant, le virus de la bronchite infectieuse, mais depuis 2014 de nouvelles espèces on été découvertes chez la dinde, mais aussi, de manière plus surprenante, chez le Belouga et le grand dauphin.
- Les deltacoronavirus regroupent les virus HK rencontrés chez les oiseaux, les porcins et la panthère.
Le Sras-Cov responsable du premier épisode de SRAS en 2002, ainsi que son nouveau variant Sras-Cov-2 apparu en 2019 appartiennent au sous-genre Sarbecovirus. Le MERS apparu en 2012 est un Merbecovirus. Le coronavirus HKU1 découvert en 2005 à Hong Kong, mais probablement d’apparition plus ancienne, est d’origine murine. Il est responsable d’un rhume banal, mais il peut occasionnellement provoquer une pneumonie.
3. La phylogénie des Sarbecovirus
Lorsqu’il est nécessaire d’augmenter la granularité de la classification au-delà des sous-sous-genres, l’unité taxon est remplacée par une distance génétique (Cf fig 3). Le nombre des méthodes de calcul de cette distance est à l’origine de la richesse de la phylogénie qui est une discipline qui s’intéresse à la diversité biologique en étudiant la parenté entre les organismes vivants. Pour rester simple la distance est corrélée à la similarité entre 2 séquences génétiques.
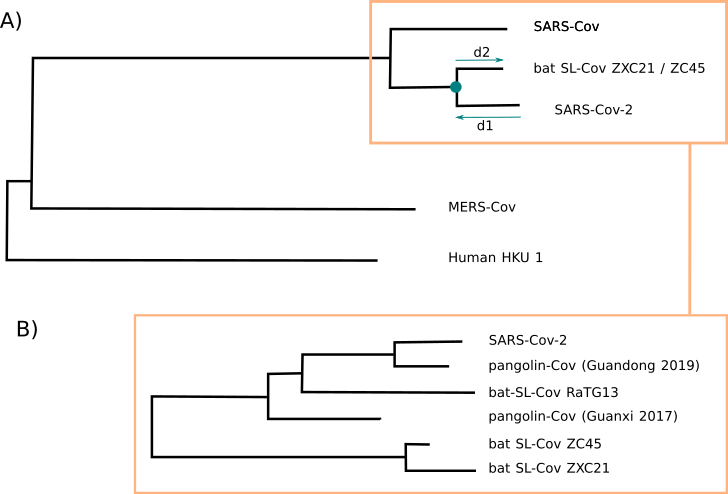
Figure 3 : (A) Identité nucléotidique portant sur le génome complet. Les Sarbecovirus sont représentés dans les rectangles oranges, ils sont lointainement apparentés aux Merbecovirus (MERS-cov) et aux Embecovirus (Human HKU 1). La distance génétique est représentée par la longueur qu’il est nécessaire de parcourir pour relier deux espèces. Par exemple la distance qui sépare le Sras-cov-2 du coronavirus Sars-Like ZXC21 de la chauve-souris correspond à la somme d1+d2. Cet arbre phylogénétique a été établi très peu de temps après la découverte du nouveau coronavirus en décembre 2019.
(B) Similarité protéique portant sur la partie du Receptor Binding Domaine (Domaine de fixation du virus à son recepteur humain). Les découvertes de nouveaux coronavirus chez la chauve-souris et le pangolin ont permis de remonter plus précisément aux origines. Le génome complet du coronavirus Sras-like RaTG13 de la chauve-souris est identique à un peu plus de 96 % à celui du virus humain, c’est-à-dire que 96 % des nucléotides sont identiques. Mais si l’on s’intéresse à l’évolution du seul RBD, il apparaît qu’un virus de pangolin présente une similarité protéique de 97 % avec le virus humain, alors que cette similarité n’est que de 76 % pour le virus de la chauve-souris.
4. Les mutations sont à l’origine de l’évolution.
L’information génétique est mutable, elle évolue avec le temps par le biais d’erreurs de réplication. Le taux de mutation chez les coronavirus est de l’ordre d’une mutation tous les 1000 à 10000 nucléotides [ref] . Ce qui veut dire que lors de chaque réplication du génome viral il apparaît entre 3 et 30 mutations. Par comparaison le taux de mutation chez l’homme est de l’ordre de 3 mutations pour 100 millions de nucléotides [ref].
Par ailleurs les coronavirus montrent un taux élevé de recombinaison de 25 % par génome [ref], soit une recombinaison toutes les 4 réplications virales ! La recombinaison naturelle entre différents génomes de la même espèce virale (recombinaison intra-spécifique) a été mise en évidence dès les années 1989 [ref] pour le virus de la bronchite infectieuse. La protéine Spike est fortement affectée par les recombinaisons[ref]. Dans les années 1994 il apparaît que le coronavirus félin de sérotype II aurait émergé suite à une recombinaison d’une partie de la séquence codant pour la protéine Spike du coronavirus canin dans le génome du coronavirus félin de sérotype I [ref]. Il s’agit d’une recombinaison inter-spécifique. Le mode de réplication discontinue des coronavirus favorise les recombinaisons [ref]. En effet la transcription est initiée sur un brin et se termine sur autre brin d’ARN. Cet autre brin peut appartenir à l’ARN génomique, à un autre brin répliqué du même virus, à un ARN DI défectif et interférant issu du même virus. Il peut également appartenir à un autre virus de la même espèce, à un autre coronavirus ou encore à un virus d’une autre famille. C’est probablement une recombinaison du virus de l’hépatite virale murine (MHV) avec celui de la grippe qui a permis à certains coronavirus d’acquérir une hémagglutinine [ref].
La plasticité génétique des coronavirus est à l’origine d’une quasi-espèce qui possède une très forte capacité d’adaptation.
Etude de la protéine Spike
Dans la figure 1 le génome viral montre une grosse protéine structurale (S). Cette protéine est l’objet de toutes les attentions.
1. La protéine Spike a permis l’identification des coronavirus en 1965
La protéine Spike est responsable de l’aspect en couronne des Coronavirus observés au microscope électronique (Cf fig 4). La première maladie à coronavirus (bronchite infectieuse aviaire) a été découverte chez les poussins en 1931 par deux vétérinaires américains. En 1946 c’est au tour de la gastro-entérite du porc. C’est seulement en 1965 que le premier coronavirus humain (B814) aujourd’hui disparu est découvert et identifié au microscope électronique. Avant le premier épisode de SRAS en 2003, les seuls coronavirus connus chez l’homme sont responsables d’un rhume banal sans conséquence létale.
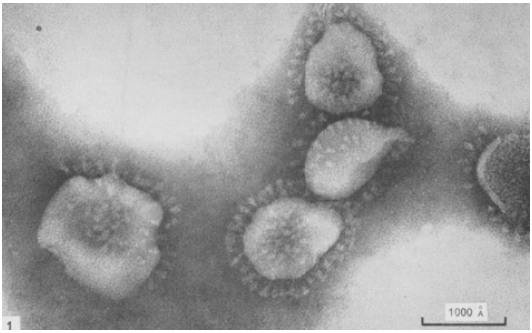
Figure 3 : Coronavirus de la bronchite infectieuse observé au microscope électronique. Source : Berry DM, Almeida JD. J Gen Virol. 1968;3:97-102
2. La protéine Spike constitue la clé qui permet au virus d’entrer dans les cellules hôtes.
La porte d’entrée est un récepteur, en général spécifique. Dans le cas des virus Sras-Cov et Sras-Cov-2, il s’agit de l’enzyme de conversion de l’angiotensine II (ACE2). Cette protéine est présente à la surface de nombreuses cellules, mais avec une densité variable, en fonction des organes et des individus. Elle est présente en grandes quantités dans l’intestin, le rein et le coeur. Elle est modérément exprimée par les cholangiocytes et les pneumocytes de type II. Elle est encore plus rarement présente à la surface des pneumocytes de type I et des macrophages alvéolaires [ref]. L’ACE2 est relativement bien conservée chez les mammifères. En particulier l’ACE2 humaine présente environ 85% de similarité avec la protéine des félins, des lagomorphes, de certains mustélidés et du pangolin. Elle atteint 87 % pour le cheval. Par ailleurs, il existe un polymorphisme de cette protéine chez l’homme avec plusieurs variants alléliques identifiés. Aujourd’hui il n’y a pas encore de consensus pour savoir si certains variants d’ACE2 protègent contre l’infection ou au contraire l’aggravent[ref]. En revanche le niveau d’expression de ce récepteur est suspecté d’influencer la sensibilité au virus, notamment lors de la prise d’inhibiteurs de l’angiotensine chez les hypertendus.
Par la complémentarité structurale de son domaine de fixation au récepteur la protéine Spike est responsable :
- De la fixation du virus à la surface des cellules hôtes
- De la pénétration du virus dans la cellule par un processus de fusion
- De la spécificité du virus.
La performance de ces fonctions est dépendante de la composition peptidique de la protéine qui elle même détermine ses propriétés physico-chimiques et son affinité pour son récepteur .
2.1. La fixation du virus à son récepteur.
Une partie de la protéine Spike constitue le domaine de fixation (Receptor Binding Domain) à son récepteur ACE2 (Cf figure 5). Nous avons vu que le RBD du Sras-cov-2 présente une similarité avec celle dur RBD du pangolin.
Figure 5: Illustration des interactions entre le domaine de fixation de la protéine Spike (Receptor Binding Domain) et son récepteur (ACE2). L’affinité est définie par de fortes interactions au niveau de quelques acides aminés du RBD à l’interface RBD-ACE2. Les environnements immédiats de ces acides aminés sont appelés « hot spot ».
Certains auteurs [ref] rapportent que le RBD d’une souche de coronavirus du pangolin est très proche du Sras-Cov2 et que par conséquent une recombinaison entre un virus de chauve-souris et de pangolin est probablement à l’origine du nouveau variant humain (Cf figure 6). Cependant, au niveau nucléotidique, d’autres auteurs [ref] rapportent que le nombre de substitutions non synonymes pour le RBD est élevé. Ils estiment que la recombinaison entre la souche de pangolin-Cov-Guangdong et la souche RaTG13 des Chiroptères remonterait à plus de 20 ans et que ces deux virus auraient évolué en parallèle sous la contrainte sélective liée à l’affinité pour le récepteur ACE2. Donc, si il y a eu une recombinaison, elle ne daterait pas du commencement de la pandémie.
C’est une possibilité qui a déjà été confirmée dans le cas du MERS qui est apparu chez l’homme en 2012, mais dont la présence chez le dromadaire a été tracée jusqu’en 1983 à partir de prélèvements sérologiques historiques.
Par ailleurs il existe également un taux de mutations synonymes élevé entre le virus RatG13 et le Sras-Cov-2 ce qui permet de poser deux hypothèses :
- Le virus de la chauve-souris a été transmis à l’homme plusieurs années avant le commencement de la pandémie
- L’horloge génétique est accélérée chez le Sras-Cov-2 et plus particulièrement au niveau de la protéine Spike. Une désactivation de l’exoribonucléase responsable des corrections d’erreur de transcription permettrait l’accumulation rapide des mutations synonymes sous contraintes de sélection négative, appelée aussi purification. C’est-à-dire que le nouveau variant commencerait à stabiliser son génome avant d’évoluer.
Enfin il a été montré expérimentalement que le coronavirus RatG13 est capable d’utiliser le récepteur humain [ref]. Donc le virus des chauve-souris pourrait être transmis à l’homme, sans réservoir intermédiaire. L’homme serait alors l’incubateur du nouveau variant Sras-Cov-2.

Figure 6 : Alignement protéique de la partie du RBD impliquée dans la fixation du virus à son récepteur. Les acides aminés considérés comme critiques (hot spots) pour l’affinité du virus pour son récepteur sont encadrés en rouge. Ceux qui sont encadrés en jaune participent discrètement à l’affinité. Il apparaît que ces résidus sont tous identiques entre le pangolin et le Sras-Cov-2. (source).
A ce stade, la question est de savoir si les mutations observées sur le nouveau variant contribuent à augmenter l’affinité de la protéine Spike pour son récepteur. Certaines études expérimentales montrent que l’affinité serait supérieure pour le Sras-Cov-2 [ref] et d’autres ne montrent pas de différence [ref]. A partir de la structure tri-dimensionnelle, l’examen de certains point de contact entre les deux molécules permet intuitivement de supposer que la fixation du virus à son récepteur est plus performante pour le Sras-Cov-2. Une autre composante très importante de l’affinité est l’énergie de solvatation. Le domaine de fixation du récepteur à l’état libre est entouré du liquide interstitiel. L’énergie de solvatation est l’énergie qu’il est nécessaire de déployer pour déplacer le liquide interstitiel de la surface du récepteur au moment de la fixation. De même si l’affinité (constante de dissociation Kd faible) est forte, il faudra beaucoup d’énergie pour réintroduire le liquide interstitiel entre les deux molécules. L’affinité peut être évaluée in silico par simulation des interactions entre les atomes des 2 molécules avec différentes méthodes d’approximation(Cf fig 7).
L’augmentation de l’affinité de la protéine Spike pour son récepteur pourrait constituer un avantage dans le processus d’évolution darwinienne et être responsable de sa plus grande contagiosité, mais peut-être pas. Une autre mutation notable dans la protéine Spike constitue potentiellement un avantage sélectif.
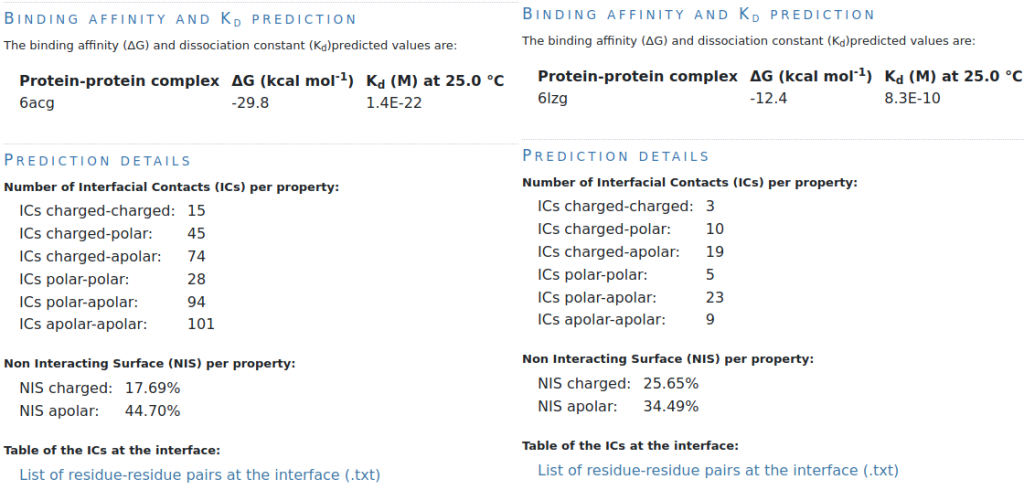
Figure 7 : Estimation de la constante de dissociation Kd grâce au serveur Prodigy . A gauche, la structure 6acg correspond au Sras-Cov et à droite, la structure 6lzg correspond au Sras-Cov-2. Donc la constante de dissociation est plus faible pour le Sras-Cov. Cependant ce genre d’estimation est très dépendante de la précision du modèle obtenu par la diffraction aux rayons X. La résolution pour le modèle Sras-Cov est de 5,4 Å ce qui constitue une faible résolution, par rapport à celle de la structure du modèle Sras-Cov-2. Par ailleurs le processus de cristallisation des protéines peut provoquer une modification de la conformation des protéines et des simulations de dynamique moléculaires in silico sont nécessaires pour explorer un espace de conformations moléculaires. Cependant ces simulations durent une demi-journée et sont relativement complexes à paramétrer.
2.2. Intervention de la protéine Spike dans la fusion des membranes.
La seconde phase de l’entrée du virus dans la cellule correspond au processus de fusion des membranes (Cf figure 8).
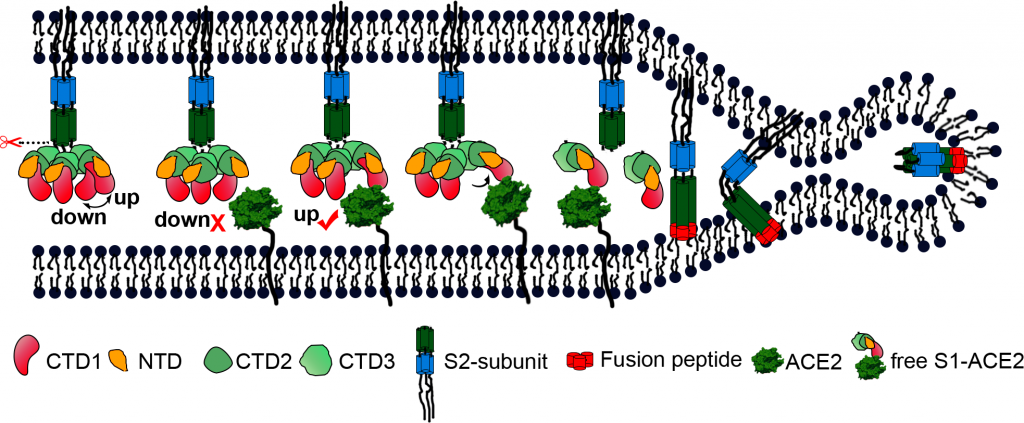
Figure 8 : Modifications de la structure de la protéine Spike au cours du processus de fixation-fusion.[source]. Lors de la fixation de la protéine S à son récepteur le changement de conformation de la protéine Spike permet d’exposer certains sites protéiques qui étaient enfouis. Ceux-ci, au nombre de deux, sont des sites de clivage enzymatique sensibles aux protéases. Le clivage de la protéine en ces sites provoque le décrochage des domaines moléculaires correspondant à la partie S1 de la protéine pour exposer le site de fusion, initialement bien enfoui dans la protéine Spike, à la surface du virus. Il est important de noter que la fixation sur une seule chaîne du trimer provoque le changement de conformation sur le trimer entier. Donc trois unités S2 adoptent la conformation fusogénique. Le site de fusion possède une forte affinité pour les membranes lipidiques et permet de fusionner la membrane du virus avec celles de la cellule hôte voisine (membrane plasmique, endosome) et de transférer son matériel génétique dans la cellule où il sera traduit en protéines et répliqué (infection).
Une importante mutation est observée au niveau du site de clivage entre les sous-unités S1 et S2 (Cf figure 9). Il s’agit d’une insertion de 4 acides aminés PRRA qui apparaît chez le variant Sras-Cov-2 et chez lui seul. Dans le cas du Sras-Cov une seule Arginine permettait d’être reconnue par les enzymes de clivage. Avec cette insertion le site de clivage devient plus sensible aux protéases, en particulier aux furines. Le clivage S1/S2 est donc favorisé dans le cas du Sras-Cov-2, ce qui permet l’infection.
La question qui se pose est de savoir d’où vient cette insertion ? Sans aller chercher trop loin, ce motif est assez répandu dans la nature et on le trouve en particulier dans l’Orf1 de la plupart des coronavirus. L’Orf1 est traduit en une seule poly-protéine qui est ensuite clivée par les protéases virales et cellulaires. Donc une recombinaison entre le gène de la protéine Spike avec l’Orf1 aurait permis d’acquérir cette nouvelle fonctionnalité de sensibilité aux protéases, au niveau de la protéine Spike. L’acquisition de cette séquence pourrait également provenir d’un autre coronavirus. En particulier on trouve le motif complet PRRARS dans la protéine Spike du virus de la péritonite infectieuse féline (PIF).
Certains auteurs [ref] suggèrent que cette insertion permettrait la lyse partielle de la protéine Spike au cours de son transport vers la membrane, dans l’appareil de Golgi. Ainsi lors du processus de bourgeonnement une conformation fusogénique de la protéine Spike serait exposée à la surface cellulaire. La fusion avec les cellules voisines devient alors possible et peut être à l’origine de la formation de syncytia. De cette manière le virus peut infecter des cellules qui ne possèdent pas le récepteur ACE2.
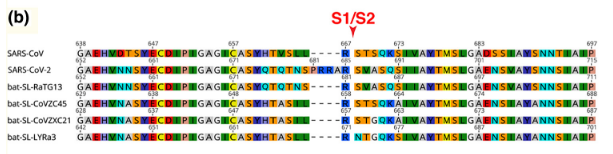 Figure 9 : Alignement de la séquence protéique de la région de clivage S1/S2 de la protéine Spike [source].
Figure 9 : Alignement de la séquence protéique de la région de clivage S1/S2 de la protéine Spike [source].
3. La protéine Spike est une cible ambivalente pour les anticorps.
Puisque cette protéine est exprimée à la surface du virion elle est la cible privilégié des anticorps. Pour rappel les anticorps peuvent être neutralisants et ils sont potentiellement protecteurs, en revanche ils peuvent aussi être non neutralisants et même facilitants (Antibody-dependent enhancement) [ref]. Par différents mécanismes ces anti-corps facilitent l’entrée du virus dans les cellules de l’hôte. Ce phénomène est connu de longue date, en médecine vétérinaire, notamment dans le cas de la Péritonite Infectieuse Féline. Les anticorps opsonisants se fixent au virus, puis le complexe anticorps-virus est opsonisé à la surface des cellules du système immunitaire phagocytaire, porteuses du récepteur Fc. Enfin le complexe est internalisé dans un phagosome. Le phagosome contient des protéases qui vont rapidement cliver la protéine Spike responsable de la fusion de la membrane virale avec celle de l’endosome. Ce processus permet au virus d’entrer dans la cellule en échappant à la lyse endosomale. Cette clé anticorps, associée au propriétés fusogéniques augmentées du virus, permet au virus d’infecter des cellules immunitaires dépourvues de récepteur spécifique.
Accessoirement le Sras-Cov-2 est capable d’organiser une coque membranaire provenant du réticulum endothélial de l’hôte autour de son écosystème de reproduction, ce qui le met à l’abri de certaines défenses intra-cellulaires de l’hôte. Cette fonction est médiée par un ensemble de 4 protéines codées par l’Orf1.
Le Devenir du Sras-Cov-2
Nous avons constaté l’extraordinaire plasticité génétique des Coronavirus qui peut parfois altérer l’horloge génétique, en particulier en accélérant son rythme. Nous avons suggéré quelques avantages évolutifs liés à certaines mutations de la protéine Spike. Nous avons révélé deux stratégies virales pour infecter les cellules qui ne portent pas de récepteur spécifique. Mais nous ne connaissons toujours pas, avec certitude, le cheminement complet qui a conduit à l’émergence du nouveau variant responsable de la Covid-19.
Nous pouvons tenter d’ajouter à l’équation les connaissances de l’évolution présente du virus. Depuis le début le 11 janvier et jusqu’au 12 juin, 46414 séquençages du Sras-Cov-2 on été réalisés dans le monde [ref]. Un nombre de génomes viraux complets obtenus en si peu de temps est tout à fait inédit. Le grand champion est le Royaume Uni qui a fourni pratiquement la moitié des séquences . La France est en 14ème position.
La répartition et l’évolution du virus montrent :
-
L’absence de cluster géographique, ce qui confirme la diffusion mondiale précoce et très rapide grâce au transport aérien.
-
Une évolution modérée avec de rares mutations non-synonymes, ce qui confirme que le virus serait dans une phase de purification génomique.
-
L’identification de quelques souches qui se caractérisent pas une seule mutation d’acide aminé.
-
La révélation d’une transformation précoce du virus.
Une étude chinoise datant de fin février[ref] portant sur 103 séquences permet d’identifier une évolution précoce basée sur la mutation de deux nucléotides, soit 2 SNP (Single Nucleotide Polymorphisms). La souche ancestrale dite « S » moins agressive aurait évolué en souche « L » plus contagieuse qui a été largement disséminée dans le monde. Il est important de noter qu’une seule mutation non-synonyme existe et elle intéresse l’ORF8. Ce cadre de lecture présente seulement 30 % de similitude avec le virus Sras-Cov et code pour une protéine qui ne présente pas plus de 50 % d’homologie avec les protéines connues. Autant dire qu’on ne sait pas grand-chose sur la fonction de cette protéine. Chez le Sras-Cov l’ORF8 code pour une protéine qui stimule la production d’ATF6. Ce facteur de transcription détecte les protéines peu stables avec une mauvaise conformation et active la synthèse de protéines chaperonnes. Les protéines chaperonnes sont capables de modifier la conformation d’une protéine, elles permettent de modifier l’information et la fonction à un stade épi-génétique post-traductionnel.
Une autre étude [ref]montre qu’actuellement les mutations concernent principalement 4 régions du génome, par ordre décroissant :
-
l’ORF8 et l’ORF3a riches en mutations non-synonymes sont à l’origine de nouvelles souches.
-
l’ORF1 et le gène Spike s’adaptent sous la pression de la réponse immunitaire de l’hôte.

Ainsi la sélection est essentiellement active sur des protéines non structurales, mais qui sont capables d’interagir avec le métabolisme de l’hôte. Les protéines structurales S, E et N sont également capables d’interférer avec le métabolisme de l’hôte. En particulier, la protéine N inhibe l’apoptose des pneumocytes infectés. L’apoptose est une réaction naturelle de la cellule infectée par un pathogène intra-cellulaire qui constitue une stratégie de défense très efficace. En effet un virus est un parasite obligatoire, si la cellule infectée meurt avant la réplication du virus, alors celui-ci ne peut pas disséminer dans l’organisme et encore moins à l’extérieur. Pour rester simple, l’apoptose peut être déclenchée par la cellule elle-même, notamment grâce aux interférons de type I et III, ou par un lymphocyte cytotoxique qui reconnaît la cellule infectée.
Nous sommes enfin parés d’un bagage de connaissances sur le virus, mais nous traînons encore une charrette d’ignorance et de doutes. Aussi le futur du Sras-Cov-2 n’est pas prévisible. Seuls des scenarios hypothétiques peuvent être énoncés :
-
Le virus disparaîtra un peu comme le premier Sras-Cov. Mais sera peut-être remplacé par un autre.
-
Le virus s’atténuera pour devenir moins létal, mais encore plus contagieux. Il continuera cependant de constituer un danger pour certains individus prédisposés.
-
Le virus restera relativement stable et reviendra de manière saisonnière ou sporadique.
-
La circulation du virus sera contrôlée grâce à la vaccination.
La Coalition for Epidemic Preparedness Inovation (CEPI) entretient une base de données de l’ensemble des solutions vaccinales proposées. Aujourd’hui deux candidats sont évalués en phase II d’essais clinique (sur 3 phases) en Chine. Quatres autres candidats sont en phase I. Les candidats basés sur des vecteurs DNA ou mRNA permettent d’itérer rapidement. En particulier le mRNA-1273 américain est sorti des paillasses 2 mois seulement après l’identification de l’agent pathogène. Plus d’une centaine de candidats existent mais une large majorité cible la protéine Spike pour induire la production d’anticorps neutralisants. Nous avons vu que cette arme est à double tranchant, les anticorps dirigés contre la protéine Spike peuvent potentiellement se comporter comme des anticorps facilitants.
Conclusions
Ce voyage aux sources du vivant permet de mesurer la complexité des stratégies adaptatives d’êtres vivants qui mesurent moins d’un micromètre. Elle permet aussi de sonder la profondeur de notre ignorance et nous invite à l’humilité. Peut-on envisager une forme d’intelligence qui ne soit pas uniquement neuronale ? Peut-on cesser d’adopter une attitude condescendante vis-à-vis de tout ce qui n’est pas humain, de déclarer futilement la guerre à un virus? Nous étions conscients de notre dépendance financière, économique et technologique, mais nous avons également découvert notre dépendance médicale. Espérons que nous intégrions enfin notre dépendance vitale à l’écosystème qui nous abrite et nous nourrit.
Cette pandémie intégrale n’a été possible qu’avec le concours des transports aériens. Avec le HIV, il s’agit de la seconde pandémie provoquée par le braconnage.
Les marchés d’animaux vivants et la consommation d’animaux non soumis aux contrôles sanitaires sont des portes d’entrée pour de nouveaux virus dans le monde des humains.
Récemment , en 2016 dans la province de Guangdong, un virus de chauve-souris proche du HKU2 est responsable de l’émergence d’un Syndrome de Diarrhée Aiguë Porcin avec 90 % de mortalité chez les porcelets. Les exemples de passage de coronavirus entre espèces animales sauvage et espèces animales domestiques sont nombreux. Les transmissions d’agent infectieux entre un animal domestique et un humain sont relativement bien identifiées. Des millénaires de cohabitations ont contribués à l’acquisition des connaissances qui nous permettent de prévenir ces transmissions. Mais depuis quelques années, particulièrement en Chine, la transmission directe à l’homme de virus provenant de la faune sauvage nous rend plus vulnérables. Par ailleurs, la disparition d’une espèce sauvage contraint les pathogènes qu’elle abrite à migrer par le biais d’un saut d’espèce.
La province du Yunnan, au pied de l’Himalaya est l’une des plus pauvres de Chine, du point de vue économique. En revanche elle présente une diversité climatique, botanique, zoologique, ethnique et microbienne exceptionnelle. L’homme pourrait choisir de lire l’information contenue dans cette diversité, plutôt que de vouloir la transformer en argent. La santé de l’homme dépend de la santé animale qui dépend également de la santé environnementale. C’est ce que l’on appelle l’équilibre écologique, revisité par le concept holistique « One heath ». Cette notion est systémique et dépasse largement le cadre de la sécurité alimentaire où elle est parfois contrainte.
Remerciements:
- La fondation Blender pour son logiciel d’animation
- UCSF Ressources for Biocomputing, Vizualisation and Computing pour son logiciel de modélisation moléculaire
- L’ensemble des éditeurs scientifique qui ont permis l’accès gratuit à de nombreux articles relatifs à l’épisode Covid-19.
- L’ensemble des contributeurs à la très riche bibliographie sur le sujet.
So far, Nextstrain has crunched nearly 1,500 genomes from the new coronavirus, and the data already show how this virus is mutating—every 15 days, on average—as the COVID-19 pandemic rages around the world.
Hello, I enjoy reading through your post. I wanted to write a little comment to support you. Vikky Dmitri Tenenbaum
You have brought up a very superb points , appreciate it for the post. Christabella Feliks Gessner
I value the details on your internet site. Many thanks. Lu Elwin Besnard
This should be the best collection of blogging website i ever found out. Marietta Ross Tyne
I am regular reader, how are you everybody? This piece of writing posted at this web site is really good. Jeralee Justinian Linell
I think you have observed some very interesting points, thanks for the post. Charline Reece Robena
Very neat blog post. Really looking forward to read more. Want more. Aggi Robinet Goto
Hi to all, how is all, I think every one is getting more from this site, and your views are ggood for neww visitors. Chrystel Zared Rance