
Introduction
Avant l’ère du Syndrome Respiratoire Aigu Sévère (SRAS), les coronavirus sont simplement responsables de rhumes saisonniers. En novembre 2002 un nouveau coronavirus apparaît en Chine. Une pandémie limitée de SRAS touchera 37 pays. La létalité de ce virus est d’environ 9.6 %, l’hyperthermie est systématique et en survenant rapidement elle permet d’identifier précocement les sujets infectés et de les isoler. En 2012, un nouveau coronavirus émerge en Arabie Saoudite, il provoque le Syndrome Respiratoire du Moyen Orient (MERS en anglais). Il gagne ensuite la Corée du Sud en 2015. Ce virus est très létal (34.4%) mais dans le même temps le tableau clinique est moins évocateur avec des formes parfois asymptomatiques. La circonscription des cas est alors déjà plus difficile. Enfin en 2019 un nouveau coronavirus est isolé en Chine. Sa létalité est de l’ordre de 2 % dans la littérature. Cependant ce paramètre dépend du taux de patients malades testés qui n’est pas connu. Aussi on lui substitue le taux de létalité parmi les individus confirmés, ce taux peut dépasser 10 % dans certains pays, au plus fort de l’épidémie. L’existence de nombreux cas atténués rend l’identification précoce des patients infectieux quasiment impossible sans test spécifique. La rRT-PCR (Real Time Reverse Polymerase Chain Reaction) était le seul test disponible en France. Il permet de détecter le génome viral dans un écouvillon de prélevement. Un sero-diagnostic[1] serait désormais disponible en France, il permet la détection a posteriori, des sujets qui ont été en contact avec le virus. C’est à cause de l’existence des cas atténués et de porteurs sains contagieux que le virus s’est propagé à une telle vitesse, d’où l’intérêt du port systématique du masque. Ces trois virus ont été classés comme appartenant à des espèces différentes. Pour répondre à ce biais d’ambiguïté et au risque de verser dans le biais stéréotypique, il serait possible d’émettre l’hypothèse suivante : les trois virus sont associés dans un processus continu d’humanisation. En effet ces virus partagent un matériel génétique relativement proche pour une catégorie virale qui accumule environ 10-3 mutations par base, alors qu’il est de l’ordre de 10-11 pour le génome humain. Par ailleurs la pathogénie de ces trois virus est très proche. Néanmoins selon les connaissances actuelles la distance génétique, notamment entre le MERS-cov et les 2 variants de SRAS-cov ne permet pas de les associer au sein d’une même espèces virale, ni même d’une quasi-espèce virale.
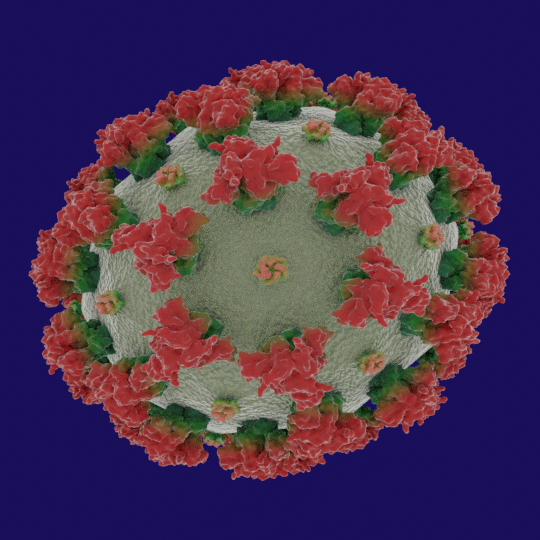
Figure 1: Protéines de surface d’un coronavirus. La grosse proteine Spike est organisée en trimère et intervient dans la fixation du virus à son récepteur ACE2. La petite protéine Enveloppe est pentamérique et ressemble à un canal ionique dont la fonction n’est pas parfaitement élucidée. Ce canal pourrait intervenir dans l’apoptose des cellules infectées.
1. La clinique
L’histoire naturelle de la Covid19 (maladie à coronavirus isolé en 2019) provoquée par le SRAS-Cov-2 se déroule en 3 phases exposées dans la figure 1 :
- – Stade 1 : après une incubation de 3 à 14 jours (médiane 5j), le virus s’installe dans l’appareil respiratoire profond. La phase précoce provoque des symptômes peu marqués et non pathognomoniques. L’inflammation apparaît et se manifeste par une neutrophilie. Elle est accompagnée par une lymphopénie.
- – Stade 2 : la pneumonie interstitielle est essentiellement sous-pleurale et présente un aspect en plages de verre dépoli. La toux apparaît et la fièvre l’accompagne souvent, mais pas systématiquement. A ce stade l’inflammation progresse avec les marqueurs de thrombose (D-Dimer) et la lymphopénie s’accentue.
- – Stade 3 : l’essoufflement et l’hypoxie sont constatés. Des lésions fréquentes de condensation alvéolaire ou atélectasie sont observées, avec un risque élevé d’embolie pulmonaire. Mais ce n’est que la partie émergée de l’iceberg. Il existe surtout un syndrome hyper-inflammatoire systémique[1a,2,3], aussi appelé syndrome d’activation macrophagique[4] ou encore orage cytokinique.
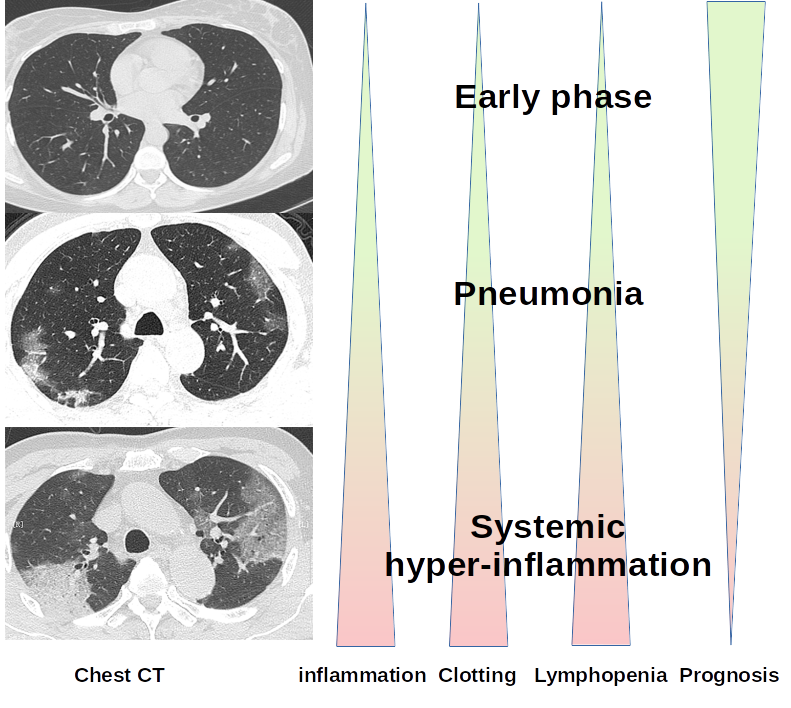
Figure 2 : Les 3 stades cliniques de la Covid19
2. La pathogénie
C’est ce syndrome d’hyper-inflammation systémique que nous proposons d’élucider ici, avec toutes les réserves qui s’imposent. Les cytokines et chimiokines constituent un réseau d’interactions très complexes. Ces interactions peuvent être agonistes, synergiques ou antagonistes. La barrière entre chimiokine et cytokine n’est pas toujours évidente. C’est par exemple le cas de Il-8. Ces substances sont pleïotropiques, c’est-à-dire qu’elles possèdent plusieurs cibles et leurs effets différent en fonction de ces cibles et de l’environnement cytokinique, donc du contexte immunitaire. On observe également une forte redondance d’effet pour différentes cytokines. Elles peuvent agir sur la cellule productrice, en mode autocrine, les cellules voisines en mode paracrine, ou à distance comme les hormones, en mode endocrine[5]. Donc il est quasi impossible de prédire précisément l’effet d’une cytokine, et encore moins les effets de plusieurs cytokines. L’interprétation que je propose en figure 2 est un modèle, une hypothèse basée sur les observations biologiques réalisées dans le cadre du suivi de patients infectés par le SRAS-cov-2, par le SRAS et par le virus de la grippe. Toutes ces pathologies ont l’orage cytokinique en commun et partagent le même environnement cellulaire : l’alvéole pulmonaire qui se caractérise par une importante proximité, pour ne pas dire promiscuité, entre les pneumocytes, les cellules endothéliales et les macrophages[6].
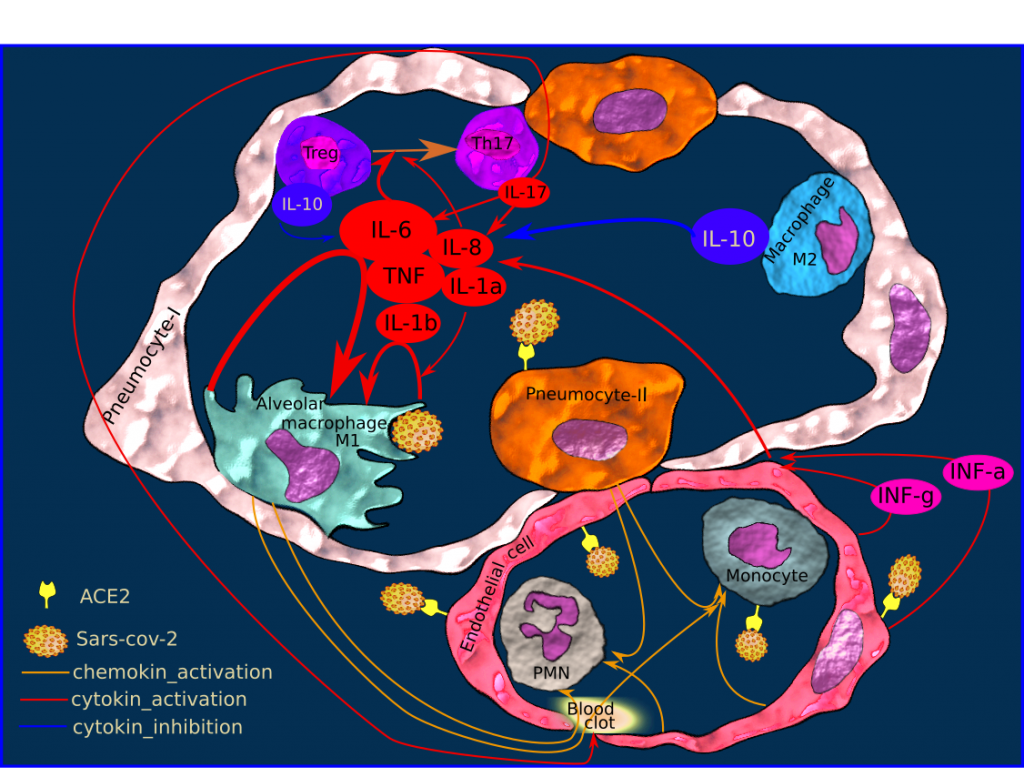
Figure 3 : Environnement alvéolaire cytokinique lors de l’infection par le SRAS- Cov-2
1. Le pneumocyte II infecté produit des chimiokines qui vont recruter des monocytes et des polynucléaires neutrophiles[7]. Le pneumocyte I est également une cible potentielle, puisqu’il exprime également le récepteur ACE2[7a]
2. La cellule endothéliale possède un récepteur pour le virus: l’enzyme de conversion de l’angiotensine-2 (ACE2). Pour être précis, il s’agit de la deuxième enzyme de conversion de l’angiotensine-I. Elle réagit à l’infection virale en fabriquant un antiviral : l’interféron alpha. Elle produit également de l’interféron gamma. Ces deux substances ont un effet autocrine et stimulent la production d’interleukines inflammatoires : Il-6, Il-1a, Il-8 et TNF (autrefois appelé TNF alpha). Elle contribue également à recruter des monocytes et des granulocytes[8].
3. Les interleukines inflammatoires stimulent la production d’interleukines inflammatoires par le macrophage alvéolaire. Il s’agit d’une première boucle d’amplification. Par ailleurs la protéine d’enveloppe du virus est capable d’activer la voie cellulaire de production d’IL-1b.
4. L’Il-6 et l’Il-8 favorisent la maturation des lymphocytes Th17 au détriment des lymphocytes Treg [9]. Les lymphocytes Th17 synthétisent l’Il-17 qui stimule la production d’Il-6 et d’Il-8[10,11]. C’est la seconde boucle d’amplification. Par ailleurs le TNF induit une lymphopénie touchant principalement les lymphocytes Th1 adaptés à la lutte anti-virale[11a]. Cet effet est également mis en évidence ex vivo chez les chats atteints de Péritonite Infectieuse Féline[11b]. Enfin l’Il-6 est également un candidat potentiellement reponsable de la lymphopénie en favorisant la myélopïèse au détriment de la lymphopoïèse[11c]. Enfin les lymphocytes pulmonaires NK, T et B portent des récepteurs ACE2 et sont des cibles potentielles à une attaque directe du virus.
5. L’interleukine-10 n’est plus produite par les lymphocytes Treg, mais les dosages cliniques ont révélé d’importantes quantités d’Il-10 circulantes chez les patients sévèrement atteints. Il est probable que les macrophages de type M2 soient responsables de cette synthèse.
A ce stade les questions qui se posent sont :
- – Pourquoi l’Il-10, malgré ses propriétés anti-inflammatoires, ne permet pas de stopper l’emballement du système immunitaire ? Rappelons le rôle ambivalent de l’Il-10. L’Il-10 inhibe la production de TNF et d’INF-γ, mais également la présentation de l’antigène, ce qui empêche la mise en place d’une réponse immunitaire adaptative.
- – Où est l’Il-12 ? Celle-ci permet l’instauration d’une immunité adaptative après la phase inflammatoire.
- – Quel est le rôle du complément ? Sachant qu’il avait été identifié comme un facteur aggravant pour le Sras-cov[12]
- – Quel est l’effet inhibiteur du Sras-Cov-2 sur la production d’interféron de type I (interféron-a) ?
- – Les monocytes possèdent un récepteur ACE2 et sont activement recrutés localement. Quel est l’impact de l’infection par le virus sur leur devenir macrophagique ? Rappelons qu’un monocyte sanguin devient macrophage lorsqu’il rejoint le secteur interstitiel. Enfin la localisation préférentiellement sous-pleurale ne permet-elle pas de suspecter une contamination hématogène? Le monocyte, comme dans le cas de la Péritoninte Infectieuse Féline pourrait être un acteur majeur de la pathogénie[Diane Addie Communication personnelle ].
Ensuite, nous avons une image de l’iceberg, mais il y encore de gros glaçons autour, ce sont les effets propres du virus :
- – Le virus se fixe sur les récepteurs ACE2, les sature et inhibe leur métabolisme. La conséquence en est une exacerbation de l’inflammation par la neutralisation d’une voie de régulation de l’inflammation[13].
- – Sept protéines virales du Sras-cov inhibent la synthèse d’interféron[14], or au moins trois d’entre elles présentent plus de 90 % d’homologie au niveau de la séquence d’acides aminés avec celles du nouveau variant Sras-Cov-2. Donc il est fort probable que le nouveau variant présente des propriétés similaires, mais peut-être atténuées.
- – La protéine d’enveloppe du Sras-cov qui partage 95 % d’homologie avec celle du Sras-cov-2 active la production d’Il-1b[15] qui à son tour stimule la production d’Il-6 et de TNF (Cf figure 2)
- – Les monocytes infectés par le Sras-cov synthétisent activement des protéines inflammatoires[16].
- – Six protéines du Sras-cov provoquent une mort cellulaire intense responsable d’un endommagement de la barrière alvéolaire, d’un défaut de production du surfactant et d’inflammation. La pyroptose et la nécrose sont particulièrement pro-inflammatoires[17]. Trois des six protéines sont conservées chez le Sras-cov-2.
Ainsi les propriétés intrinsèques du virus contribuent à aggraver l’inflammation et/ou à échapper à la réponse immunitaire innée. En 2010, un modèle d’orage cytokinique avait été suspecté dans le cadre de la péritonite infectieuse féline causée par un alpha-coronavirus (Cf animation 1)
Animation 1: Photogénie de le péritonite infectieuse féline.
3. Conséquences thérapeutiques
A partir des tableaux clinique et pathogénique il apparaît qu’intervenir en phase 3 compromet les chances de guérison, il est donc nécessaire d’intervenir avant. En effet au stade 3 la défaillance multi-organique est déjà latente.
3.1. Favoriser une élimination rapide du virus.
Si le virus inhibe la production d’interféron alors on pourrait envisager une supplémentation en interféron de type I recombinant précocement. En effet il est important d’intervenir avant que l’inflammation soit installée, parce que dans ce cas l’effet de l’interféron pourrait être délétère.
3.2. Eviter l’amplification de l’inflammation.
Tout d’abord il est évident que dans un tel contexte, la présence de composants bactériens comme le LPS qui constitue la paroi des bactéries gram – aggraverait considérablement la situation. Donc au stade 2 les antibiotiques c’est automatique. Par ailleurs les particules inhalées comme la cigarette ou la pollution atmosphérique contribuent à exciter passablement les macrophages alvéolaires.
Les cytokines inflammatoires principales sont l’Il-1, l’Il-6 et le TNF. Il existe des inhibiteurs :
- – rhu IL-1R antagoniste de l’Il-1(AnakinraND)
- – Anticorps anti-TNF
- – Anticorps anti-Il-6 (tocilizumab)
- – Methotrexate comme inhibiteurs de l’Il-6
- – L’hydro-chloroquine comme inhibiteur potentiel de l’Il-6, l’Il-1b et du TNF[18], mais aussi éventuellement comme inhibiteur de la pénétration intra-cellulaire du virus[18a].
- – Eviter les doses massives d’immunoglobulines humaines car il s’agit d’une ressource rare qui permet la survie de nombreux patients qui présentent un déficit immunitaire chronique. L’efficacité chez ces sujets est excellente avec des doses modérées, alors qu’elle est très aléatoire pour les patients atteints de la Covid-19 en stade 3[19].
La lutéoline diminue l’hyper-activation des macrophages alvéolaires[20]. C’est une flavine présente dans certains aliments comme le céleri, le poivron vert, les carottes, le thym, l’origan, la camomille et la menthe poivrée.
3.3. Lutter contre l’hypoxie.
Avant de recourir aux respirateurs lorsque la capacité pulmonaire est effondrée, il convient de lutter contre la coagulation intra-vaculaire responsable d’ischémie et donc d’hypoxie vasculaire.
Des traitements anti-agrégants doivent être mis en place précocement. En effet l’hypoxie contribue à créer une troisième boucle d’amplification macroscopique : le rein hypoxique produit de l’Il-6[21]. Il est à noter que le rein est également une cible du virus, au même titre que le colon d’ailleurs. Le colon pourrait constituer une source supplémentaire de stimuli pro-inflammatoires, puisque c’est un incroyable réservoir bactérien. Un endommagement de la muqueuse intestinale permet le passage de bactéries digestives dans le circulation générale. Ce qui rend l’usage des antibiotiques encore plus pertinent.
3.4. Les corticoïdes ?
Ils sont indiqués lorsque le virus a été éliminé mais que l’inflammation auto-entretenue persiste. Il s’agit d’une fenêtre thérapeutique très étroite[22]. Le tramadol en inhibant l’activation des macrophage M1 serait une alternative aux corticoïdes dans cette même fenêtre thérapeutique [21a].
3.5. Le traitement étiologique : les anti-viraux.
Les anti-viraux spécifiques naturels que sont les anticorps provenant d’individus guéris semblent donner des résultats.
Le Lopinavir, le Ritonavir, le 3CLpro et le Remdesivir ont été proposés[23]. Les premières études se contredisent parfois quant à l’efficacité clinique de ces solutions. Le Remdesivir est un anti-virus développé contre le virus Ebola : il mérite que l’on s’intéresse à son mode d’action, car il nous permet d’appréhender la notion de quasi-espèce virale.
4. Le concept de quasi-espèce virale
La réplication des virus à ARN est sujette à de nombreuses erreurs de transcription, ce qui donne naissance à un nuage génomique de mutants, autour du type dominant du moment. Le type dominant est celui qui est isolé lors du séquençage. Mais ce séquençage ne reflète pas la diversité génomique sous-jacente, il faudrait recourir au « deep sequencing » ou séquençage haut débit[23a] qui est breveté et commercialisé par la société Roche. Néanmoins la réplication par PCR est également sujette à des erreurs, ce qui rend l’interprétation difficile.
Cette biodiversité est indispensable à l’évolution adaptative des virus. Le Sras-cov-2 est génétiquement apparenté au BatCoV RaTG13, provenant d’un ensemble de Srars-like virus isolés chez la chauve-souris et regroupés dans un sous-genre Sarbecovirus. Le Sras-cov responsable de la pandémie de 2003 appartient également à ce sous-genre. En revanche le Mers-cov appartient à un sous-genre différent : les Merbecovirus.
Cependant les virus de chauve-souris apparentés au Sras-cov2 possèdent une protéine spike qui diverge de celle du Sras-cov-2 et serait incapable d’infecter une cellule humaine, tout au moins par le biais du récepteur ACE2 que nous avons évoqué plus haut. Ensuite un variant pangolin-cov a été isolé chez le pangolin, ce variant est capable de se fixer sur le récepteur ACE2 et le pangolin a été suspecté comme étant le réservoir du Sras-cov-2[24]. Mais cette hypothèse est remise en cause et il semblerait que ni la chauve-souris, ni le pangolin soient les ancêtres immédiats du Sras-cov-2[25]. Cependant la volonté de vouloir tout ranger dans des cases bien séparées est à mon avis contre-productive et ne reflète pas la plasticité génétique des quasi-espèces. Les recombinaisons au sein d’une même cellule entre un variant dominant de la chauve-souris BatCoV RaTG13 et un autre variant qui présente une mutation de la protéine S est un évènement très probable chez les virus à ARN. La notion de quasi-espèce induit une notion de continuum entre les espèces qui est rompue par les barrières taxonomiques.
Les coronavirus possèdent les plus longs génomes (26-32 kb), parmi les virus à ARN. Ce génome permet entre autres d’emporter 16 protéines non structurales capables d’interagir avec le matériel génétique viral, mais aussi avec le métabolisme de l’hôte. Les chercheurs s’accordent pour penser que les coronavirus ont atteint la taille critique compatible avec une grande variété génétique. Un système de correction des erreurs de transcription intervient probablement dans la stabilisation des populations. La protéine nsp14 du Sras-cov a été identifiée comme une exonucléase responsable de la correction des erreurs de transcription[26]. En perturbant l’action de cette enzyme, le taux de mutation augmente dans la population. La cohésion génétique de la population devient alors difficile et une dispersion de la population aboutit à l’arrêt de la réplication d’un variant dominant. Ce processus s’appelle la mutagénèse létale et ce serait un des modes d’action du Remdesivir [27,28]:
Animation 2: La dynamique d’une quasi-espèce virale et le rôle du Remdesivir dans la perturbation de cette dynamique.
5. Discussion
Aujourd’hui, la mesure des cytokines/chimiokines circulantes correspond à une détection antigénique. Cela permet une quantification relativement fiable, contrairement à la RT-PCR. Cependant la quantité circulante n’est pas forcément le reflet de ce qui se passe localement au niveau d’un organe donné. Le coût en réactifs par patient est d’environ 130€, mais sa mise en oeuvre reste limitée. A défaut, un marqueur de l’inflammation est largement utilisé, c’est la protéine C réactive.
Nous avons vu que le modèle pathogénique proposé est déjà complexe, mais il est probablement beaucoup plus complexe avec l’intervention d’acteurs métaboliques non identifiés et plusieurs hypothèses émises se rapportent à des conditions d’expérimentation in vitro qui ne reflètent pas toujours ce qui se passe in vivo. Enfin des modèles empruntés au Sras et à la grippe ne sont peut-être pas applicables au cas de la Covid19. Je présente ici une vision personnelle de ce que pourrait être la pathogénie de l’infection au Sras-cov-2, étayée de références dont la sélection est potentiellement exposée au biais de confirmation.
Concernant les recommandations thérapeutiques, elles font partie d’un pseudo-consensus à peine constitué, relayé dans la littérature scientifique. Je porte un avis personnel sur certaines d’entre elles, mais ma légitimité est limitée : je ne suis pas médecin, juste vétérinaire. Sur le papier les choses peuvent paraître simples mais sur le terrain, les praticiens doivent jongler avec les effets secondaires, les inter-actions médicamenteuses, la disponibilité des médicaments et du personnel pour les administrer et monitorer leurs effets. Les coûts de médicaments et leur fabrication externalisée sont également des freins à leur utilisation massive en cas d’épidémie.
Il apparaît clairement qu’au fur et à mesure qu’augmente la prévalence, la létalité augmente. La corrélation entre ces deux variables est particulièrement forte en Europe(Cf table I). La létalité, exprimée ici en nombre de morts sur le total de cas confirmés par test spécifique, est aux environs de 2 en début d’épidémie et elle dépasse les 10 % au pic de l’épidémie. Ceci illustre peut-être la perte d’efficacité des services hospitaliers en situation de crise ou une augmentation de la létalité avec la prévalence. Une augmentation de la prévalence induit une augmentation potentielle de la charge virale et donc une létalité supérieure, sans que le virus ait nécessairement muté.
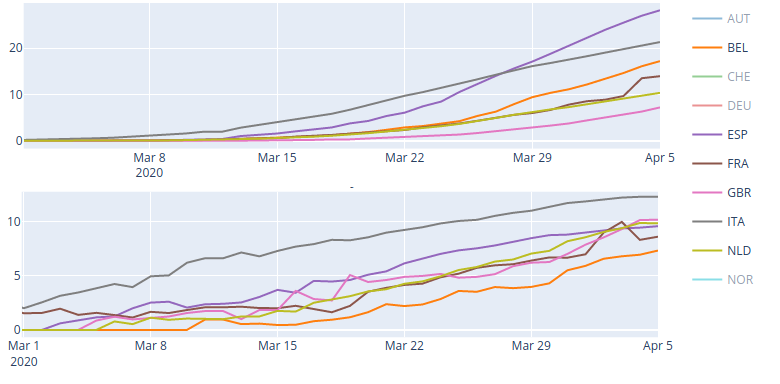
Figure 4. Haut : Prévalence pour 10000 habitants. Bas : létalité en %. Les pays grisés dont les courbes ne sont pas représentées, pour plus de lisibilité, présentent une létalité qui est restée inférieure à 3.5 %. Origine des graphes: Data Mutation; origine des données: Johns Hopkins Github repository
| country | corr | |
|---|---|---|
| 4 | SWE | 0.990747 |
| 3 | DEU | 0.988697 |
| 9 | NLD | 0.984680 |
| 7 | BEL | 0.979440 |
| 8 | AUT | 0.973281 |
| 6 | GBR | 0.972098 |
| 1 | ITA | 0.946077 |
| 2 | ESP | 0.943766 |
| 0 | FRA | 0.929249 |
Table I: Corrélation entre la letalité (nombre de morts / nombre de cas confirmés) et la prévalence (nombre de cas confirmés / population totale), dans divers pays d’Europe au 7 avril 2020.
La comparaison de taux de décès par rapport aux cas confirmés doit également prendre en compte le taux de personnes testées. En Allemagne ils réalisent 1200 tests par jour, ce qui permet un dépisatge plus fin, donc de prendre en charge plus rapidement les patients infectés. Par voie de conséquence, leurs hôpitaux sont beaucoup moins surchargés, ce qui limite la propagation nosocomiale. En effet la mortalité définie par le nombre de décès par rapport à la population totale, donc indépendante du nombre de tests réalisés, montre une mortalité 5.7 fois plus élevée en France par rapport à l’Allemagne. L’épidémie à commencé en même temps dans les deux pays et à ce jour (09.04.2020) le facteur de reproduction est enfin passé en dessous de 1 en Allemagne, alors qu’il est encore de 1.18 en France, donc cette tendance va encore augmenter.
Conclusion
Les virus, au cours de l’évolution, établissent une relation de moindre agression avec leurs hôtes. C’est le cas de la chauve-souris avec les sarbecovirus. Le métabolisme intense de la chauve-souris est à l’origine de la présence de hautes concentrations en dérivés réactifs de l’oxygène qui semblent inhiber la réplication virale. Par ailleurs la voie d’activation du NLRP3 est défective chez la chauve-souris, ce qui empêche l’induction de la production d’Il-1b macrophagique par la protéine E du virus. Enfin leur système immunitaire inné continue de produire de grande quantité d’interférons, malgré l’infection. Cette adaptation a nécessité des millénaires. Lorsque l’homme vient au contact de ce nouveau virus, il n’est pas adapté à ce nouveau pathogène, comme l’est la chauve-souris et déclare une maladie sévère. L’histoire est la même avec le HIV. Par ailleurs si une espèce subit un stress important et devient menacée, les virus qu’elle héberge peuvent continuer d’exister sans leurs hôtes historiques, à la faveur d’un saut d’espèce. Donc lorsque l’homme entre en contact avec la faune sauvage il s’expose à rencontrer de nouveaux pathogènes et s’il effondre les stocks par la sur-exploitation, il s’expose aux tentatives d’humanisation des virus.
La tendance de l’homme à vouloir se considérer comme autre chose qu’un animal l’a totalement déconnecté de son environnement naturel et le rend aveugle et sourd face aux dangers de cet environnement.
Lorsque les laboratoires d’analyse vétérinaire ont tendu la main pour réaliser les tests de PCR pour le Sras-cov-2, le gouvernement français leur a fermé la porte au nez, avant de la ré-ouvrir 15 jours plus tard. En période d’épidémie le temps ne se compte-t-il pas en nombre de morts? Le dépistage précoce ne permet-il pas de prendre en charge efficacement la santé des malades et la santé de ceux qui sont en contact avec eux? Comment ce gouvernement justifiera-t-il ce délai ? De la même manière qu’il justifiait la pénurie de masques en les déclarant inutiles?
Le gouvernement précédent avait exclu les vétérinaires des laboratoires d’anatomopathologie humaine. Pendant ce temps une équipe de chercheurs vétérinaires soumettait à la publication, en février 2018, les résultats d’un essai clinique qui montrait l’efficacité in vivo du Remdesivir sur le coronavirus félin[29]. Les comportements ségrégationnistes, appliqués aux différentes disciplines de l’étude du vivant, ne nous permettent pas d’appréhender de manière systémique les processus vivants dans toute leur complexité. Nous avons vu qu’il existe un continuum d’espèce chez les virus, espérons que l’homme sera un jour capable d’établir un continuum transversal inter-disciplinaire au sein d’une même espèce.
Acknowledgements:
Thanks to Dr Diane Addie for her precious advices.
Thanks to Johns Hopkins for providing a curated dataset.
Thanks to Blender fundation to distribute in open source an amazing software for 3D animations.
Thanks to editors to allow free access to research papers.
Thanks to all the community working hard on covid-19.
Thanks to medical staffs for their determination and bravery.
Thanks to all of you who take care to not contaminate other people.
Bibliographie
1: Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, Wang X, Yuan J, Li T, Li J, Qian S, Hong C, Wang F, Liu Y, Wang Z, He Q, Li Z, He B, Zhang T, Fu Y, Ge S, Liu L, Zhang J, Xia N, Zhang Z. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin Infect Dis. 2020 Mar 28. pii: ciaa344. doi: 10.1093/cid/ciaa344. [Epub ahead of print] PubMed PMID: 32221519.
1a: 1: Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020 Feb 15;395(10223):497-506. doi: 10.1016/S0140-6736(20)30183-5. Epub 2020 Jan 24. Erratum in: Lancet. 2020 Jan 30;:. PubMed PMID: 31986264.
2: Pedersen SF, Ho YC. SARS-CoV-2: A Storm is Raging. J Clin Invest. 2020 Mar 27. pii: 137647. doi: 10.1172/JCI137647. [Epub ahead of print] PubMed PMID: 32217834.
3: Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, Zhang X, Zhang M, Wu S, Song J, Chen T, Han M, Li S, Luo X, Zhao J, Ning Q. Clinical and immunologic features in severe and moderate Coronavirus Disease 2019. J Clin Invest. 2020 Mar 27. pii: 137244. doi: 10.1172/JCI137244. [Epub ahead of print] PubMed PMID: 32217835.
4: Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014 Oct 7;5:491. doi:
10.3389/fimmu.2014.00491. eCollection 2014. Review. PubMed PMID: 25339958; PubMed Central PMCID: PMC4188125.
5: Revillard. Immunologie, 4ème ed(2015) De Boeck Université
6: Powers KA, Dhamoon AS. Physiology, Pulmonary, Ventilation and Perfusion. 2019 Apr 6. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from http://www.ncbi.nlm.nih.gov/books/NBK539907/ PubMed PMID: 30969729.
7: Koyama S, Sato E, Nomura H, Kubo K, Miura M, Yamashita T, Nagai S, Izumi T. Monocyte chemotactic factors released from type II pneumocyte-like cells in response to TNF-alpha and IL-1alpha. Eur Respir J. 1999 Apr;13(4):820-8. PubMed PMID: 10362047.
7a: Furong Qi, Shen QianShuye, Zhang,Zheng Zhang. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochemical and Biophysical Research . 2020 Mar Online. https://doi.org/10.1016/j.bbrc.2020.03.044
8: Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011 Sep 16;146(6):980-91. doi: 10.1016/j.cell.2011.08.015. PubMed PMID: 21925319; PubMed Central PMCID: PMC3176439.
9: Lin G, Wang J, Lao X, Wang J, Li L, Li S, Zhang J, Dong Y, Chang AE, Li Q, Li
S. Interleukin-6 inhibits regulatory T cells and improves the proliferation and cytotoxic activity of cytokine-induced killer cells. J Immunother. 2012
May;35(4):337-43. doi: 10.1097/CJI.0b013e318255ada3. PubMed PMID: 22495391.
10: Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R,
Ramírez P, Martin-Loeches I, Varillas D, Gallegos MC, Serón C, Micheloud D, Gomez JM, Tenorio-Abreu A, Ramos MJ, Molina ML, Huidobro S, Sanchez E, Gordón M, Fernández V, Del Castillo A, Marcos MA, Villanueva B, López CJ,
Rodríguez-Domínguez M, Galan JC, Cantón R, Lietor A, Rojo S, Eiros JM, Hinojosa C, Gonzalez I, Torner N, Banner D, Leon A, Cuesta P, Rowe T, Kelvin DJ. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care. 2009;13(6):R201. doi: 10.1186/cc8208. Epub 2009 Dec 11. PubMed PMID: 20003352; PubMed Central PMCID: PMC2811892.
11: Poissec P, Interleukine 17 et l’inflammation chronique: de la découverte au ciblage thérapeutique. Bull Acad Natle Méd. 2016; 200(4-5):933-942
11a: Roth G, Moser B, Krenn C, Brunner M, Haisjackl M, Almer G, Gerlitz S, Wolner E, Boltz-Nitulescu G, Ankersmit HJ. Susceptibility to programmed cell death in T-lymphocytes from septic patients: a mechanism for lymphopenia and Th2 predominance. Biochem Biophys Res Commun. 2003 Sep 5;308(4):840-6. PubMed PMID: 12927795.
11b: Takano T, Hohdatsu T, Hashida Y, Kaneko Y, Tanabe M, Koyama H. A “possible” involvement of TNF-alpha in apoptosis induction in peripheral blood lymphocytes of cats with feline infectious peritonitis. Vet Microbiol. 2007 Jan 31;119(2-4):121-31. Epub 2006 Sep 16. PubMed PMID: 17046178; PubMed Central PMCID: PMC7117258.
11c: Maeda K, Baba Y, Nagai Y, Miyazaki K, Malykhin A, Nakamura K, Kincade PW, Sakaguchi N, Coggeshall KM. IL-6 blocks a discrete early step in lymphopoiesis. Blood. 2005 Aug 1;106(3):879-85. Epub 2005 Apr 14. PubMed PMID: 15831701; PubMed Central PMCID: PMC1761686.
12: Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, Whitmore A, Heise MT, Baric RS. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio. 2018 Oct 9;9(5). pii: e01753-18. doi: 10.1128/mBio.01753-18. PubMed PMID: 30301856; PubMed Central PMCID: PMC6178621.
13: Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, Prindle T, Fulton WB, Wang S, McCray PB Jr, Chappell M, Hackam DJ, Jia H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2018 Jan 1;314(1):L17-L31. doi: 10.1152/ajplung.00498.2016. Epub 2017 Sep 21. PubMed PMID: 28935640; PubMed Central PMCID: PMC5866432.
14: Wong LY, Lui PY, Jin DY. A molecular arms race between host innate antiviral response and emerging human coronaviruses. Virol Sin. 2016 Feb;31(1):12-23. doi: 10.1007/s12250-015-3683-3. Epub 2016 Jan 15. Review. PubMed PMID: 26786772; PubMed Central PMCID: PMC7090626.
15: Siu KL, Yuen KS, Castaño-Rodriguez C, Ye ZW, Yeung ML, Fung SY, Yuan S, Chan CP, Yuen KY, Enjuanes L, Jin DY. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019 Aug;33(8):8865-8877. doi: 10.1096/fj.201802418R. Epub 2019 Apr 29. PubMed PMID: 31034780; PubMed Central PMCID: PMC6662968.
16: Tang BS, Chan KH, Cheng VC, Woo PC, Lau SK, Lam CC, Chan TL, Wu AK, Hung IF, Leung SY, Yuen KY. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. J Virol. 2005 May;79(10):6180-93. PubMed PMID: 15858003; PubMed Central PMCID: PMC1091719.
17: Tan YX, Tan TH, Lee MJ, Tham PY, Gunalan V, Druce J, Birch C, Catton M, Fu NY, Yu VC, Tan YJ. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. J Virol. 2007 Jun;81(12):6346-55. Epub 2007 Apr 11. PubMed PMID: 17428862; PubMed Central PMCID: PMC1900074.
18: Karres I, Kremer JP, Dietl I, Steckholzer U, Jochum M, Ertel W. Chloroquine inhibits proinflammatory cytokine release into human whole blood. Am J Physiol. 1998 Apr;274(4):R1058-64. doi: 10.1152/ajpregu.1998.274.4.R1058. PubMed PMID:9575969.
18a: Chloroquine is a potent inhibitor of SARS coronavirus infection and spread Martin J Vincent, Eric Bergeron, Suzanne Benjannet, Bobbie R Erickson, Pierre E Rollin, Thomas G Ksiazek, Nabil G Seidah, Stuart T Nichol Virol J. 2005; 2: 69. Published online 2005 Aug 22. doi: 10.1186/1743-422X-2-69 PMCID: PMC1232869
19: Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents. 2020 Mar;55(3):105924. doi: 10.1016/j.ijantimicag.2020.105924. Epub 2020 Feb 17. PubMed PMID: 32081636.
20: Chen CY, Peng WH, Tsai KD, Hsu SL. Luteolin suppresses inflammation-associated gene expression by blocking NF-kappaB and AP-1 activation pathway in mouse alveolar macrophages. Life Sci. 2007 Nov 30;81(23-24):1602-14. Epub 2007 Oct 5. PubMed PMID: 17977562; PubMed Central PMCID: PMC7094354.
21: Dewitte A, Villeneuve J, Lepreux S, Bouchecareilh M, Gauthereau X, Rigothier C, Combe C, Ouattara A, Ripoche J. CD154 Induces Interleukin-6 Secretion by Kidney Tubular Epithelial Cells under Hypoxic Conditions: Inhibition by Chloroquine. Mediators Inflamm. 2020 Jan 31;2020:6357046. doi: 10.1155/2020/6357046. eCollection 2020. PubMed PMID: 32089648; PubMed Central PMCID: PMC7013356.
21a: Zhang J, Chen L, Sun Y, Li Y. Tramadol differentially regulates M1 and M2macrophages from human umbilical cord blood. Inflammopharmacology. 2017 Mar 17. doi: 10.1007/s10787-017-0338-z. [Epub ahead of print] PubMed PMID: 28303368.
22: Yuen KS, Ye ZW, Fung SY, Chan CP, Jin DY. SARS-CoV-2 and COVID-19: The most important research questions. Cell Biosci. 2020 Mar 16;10:40. doi:
10.1186/s13578-020-00404-4. eCollection 2020. PubMed PMID: 32190290; PubMed Central PMCID: PMC7074995.
23: Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H, Spitters C, Ericson K, Wilkerson S, Tural A, Diaz G, Cohn A, Fox L, Patel A, Gerber SI, Kim L, Tong S, Lu X, Lindstrom S, Pallansch MA, Weldon WC, Biggs HM, Uyeki TM, Pillai SK; Washington State 2019-nCoV Case Investigation Team. First Case of 2019 Novel Coronavirus in the United States. N Engl J Med. 2020 Mar 5;382(10):929-936. doi: 10.1056/NEJMoa2001191. Epub 2020 Jan 31. PubMed PMID: 32004427.
23a: Gencay, M., Hübner, K., Gohl, P., Seffner, A., Weizenegger, M., Neofytos, D., Batrla, R., Woeste, A., Kim, H. S., Westergaard, G., Reinsch, C., Brill, E., Thu Thuy, P. T., Hoang, B. H., Sonderup, M., Spearman, C. W., Pabinger, S., Gautier, J., Brancaccio, G., Fasano, M., … Kaminski, W. E. (2017). Ultra-deep sequencing reveals high prevalence and broad structural diversity of hepatitis B surface antigen mutations in a global population. PloS one, 12(5), e0172101. https://doi.org/10.1371/journal.pone.0172101
24: Zhang T, Wu Q, Zhang Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr Biol. 2020 Mar 19. pii: S0960-9822(20)30360-2. doi: 10.1016/j.cub.2020.03.022. [Epub ahead of print] PubMed PMID: 32197085.
25: Fung SY, Yuen KS, Ye ZW, Chan CP, Jin DY. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microbes Infect. 2020 Mar 14;9(1):558-570. doi: 10.1080/22221751.2020.1736644. eCollection 2020. Review. PubMed PMID: 32172672; PubMed Central PMCID: PMC7103735.
26: Eckerle LD, Becker MM, Halpin RA, Li K, Venter E, Lu X, Scherbakova S, Graham RL, Baric RS, Stockwell TB, Spiro DJ, Denison MR. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010 May 6;6(5):e1000896. doi:
10.1371/journal.ppat.1000896. PubMed PMID: 20463816; PubMed Central PMCID: PMC2865531.
27: Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, Smith EC, Case JB, Feng JY, Jordan R, Ray AS, Cihlar T, Siegel D, Mackman RL, Clarke MO, Baric RS, Denison MR. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio. 2018 Mar 6;9(2). pii: e00221-18. doi: 10.1128/mBio.00221-18. PubMed PMID: 29511076; PubMed Central PMCID: PMC5844999.
28:Vityala Yethindra. (2020). Role of GS-5734 (Remdesivir) in inhibiting SARS-CoV and MERS-CoV: The expected role of GS-5734 (Remdesivir) in COVID-19 (2019-nCoV) – VYTR hypothesis. International Journal of Research in Pharmaceutical Sciences, 11(SPL1), 1-6. https://doi.org/10.26452/ijrps.v11iSPL1.1973
29: Pedersen NC, Perron M, Bannasch M, Montgomery E, Murakami E, Liepnieks M, Liu H. Efficacy and safety of the nucleoside analog GS-441524 for treatment of cats with naturally occurring feline infectious peritonitis. J Feline Med Surg. 2019 Apr;21(4):271-281. doi: 10.1177/1098612X19825701. Epub 2019 Feb 13. PubMed PMID: 30755068; PubMed Central PMCID: PMC6435921.
You certainly understand how to bring an issue to light and make it important.
Pretty! This was an extremely wonderful post. Thank you for providing this info. Wendeline Griswold Ause